I have a genome, you have a genome. We don’t know what it contains.
Our understanding of how the genome works has limitations, but there are also a lot of really useful things that we do understand, or are able to predict once we know the contents of our genes. This capability is only going to improve.
Some companies offer genome analysis services, most notably ancestry estimation and polygenic scores calculation for various traits. However, I’ve found that:
- They are of limited interest. I’d like to design and perform my own types of analysis on my genome.
- I’d like not to have to rely on a commercial third party for these analysis.
- Personal genomics is too important not to be accessible by everyone, open-source.
From there came the idea of personal genomics software than it open source, that can be easily self-hosted, and that can be expanded by the community. It should ingest genetic information, perform desired analysis, and return the results in an easy to digest way in a web browser.
So, back in 2025, I decided to have my genome fully sequenced and to use the data to start the development of self.dna. Recently, this project acquired momentum when it was selected for the BIOMICS hackathon, and I thought this would be a good moment to stop an reflect on its possible future.
Project overview
self.dna is a personal genomics app. Differently from commercial alternatives it is open-source and self-hosted, ensuring users have full control over their genetic data and the way they are processed.
It adopts a modular design that allows the community to develop custom analysis modules. This makes it possible to unlock new insights based on ever advancing capabilities in genome analysis. At startup, the app will only load the modules the user needs.
Deploying self.dna involves cloning a GitHub repository to build the Docker container where the app will run:
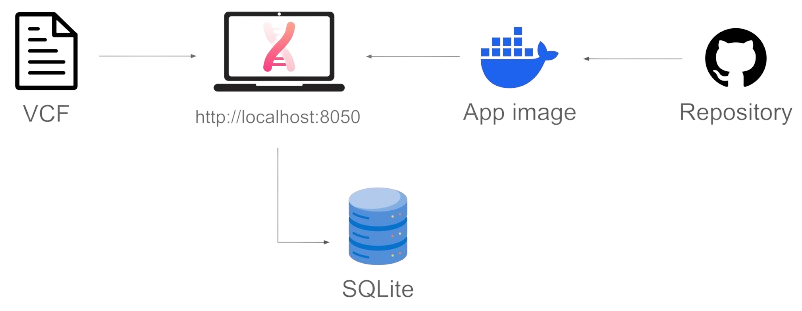
Currently, self.dna accepts the upload of a Variant Call Format (VCF) file. The VCF is stored and used to build a relational database, which can be read and written by the analysis modules. Such database persists on disk and can be ported to different instances of the app.
Hackaton achievements
We made significant progress in transforming self.dna from a simple proof of concept demo, based on a single script, into a maintainable, community-driven piece of software. The main changes involved:
-
Fast input ingest: Better support for large uploads, such as a human genome, with better utilization of the available cores.
-
Variants reference annotation: Core module to add variant annotation information from the GENCODE project. This populates the annotations table of the persistent database, to be used in many downstream analysis.
-
Modular architecture: Dynamic discovery of the analysis modules present at startup, determining the layout of the dashboard.
- Each valid module will cause the creation of a tab in the dashboard
- Each module is a Class
- Containing the attribute
tab_name - And the method
render()
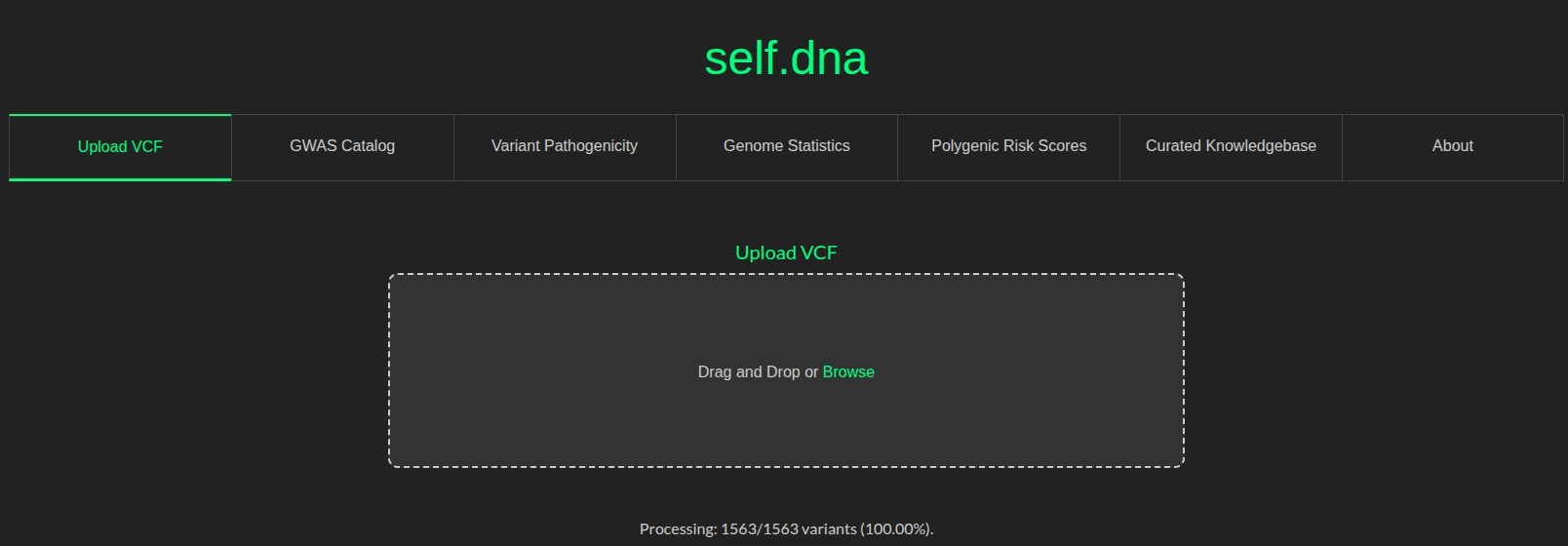
The following is a screenshot of the self.dna dashboard displaying 5 tabs/functionalities, in addition to the default tabs “Upload VCF” and “About”.
TODO
This hackaton generated a strong momentum in the development of this project. Natural next steps to finalize the work will be:
- Merge branches and refine the main codebase
- Write community guidelines and documentation for development of the analysis modules
- Write more core modules (e.g. non-sense variant mapping, ClinVar analysis)
- Consider port in WebAssembly?
Acknowledgements
My great team members: Alistair Rust¹, Erin Chung², Filipe Rodrigues³, Pedro Suzano⁴
- EMBL-EBI, Hinxton, UK
- EMBL, Heidelberg, Germany
- AccelBio, Lisbon, Portugal
- Universidade de Lisboa, Lisbon, Portugal